2026.04.16
- 医療機器物流
医療機器製造プロセスにおけるPDCAの"D"の最適化― 文書化・識別・リスク・バリデーションの実務整理

医療機器の品質マネジメントはPDCA(PLAN-DO-CHECK-ACT *)の考え方を基盤に運用されます。
*「PLAN-DO-CHECK-ACT」:以下の総称を指す。
「PLAN」 目的・目標のための手順を定める
「DO」 手順通りに実行する
「CHECK」 結果が手順通りかを評価する
「ACT」 必要に応じ、改善・標準化
この循環が機能して初めて、品質マネジメントシステムは「変動に強く、再現性があり、説明可能」な状態が維持できます。医療機器の製造は、PDCAの"D"に位置付けられます。しかし、単なる"DO(実行)した"だけでは不十分で、意図した品質を恒常的に作り出す機能を保持しなければいけません。 今回はこの"D"に焦点をあて「文書化」、「識別」、「工程リスク」、「プロセスバリデーション」という4つの観点から、実務への落とし込みを整理します。
ページ目次
医療機器の製造における文書化のポイント|工程の再現性の担保
医療機器の製造工程は、文書化・実施・維持・記録で裏付けられた、"管理されたD"でなければなりません。工程の目的・手順・判定・記録・資源(設備/測定機器/要員/作業環境)を一体設計し、最新版の文書が現場で即参照できることが前提です。
ISO13485における「製造およびサービス提供の管理(7.5)」では、医療機器の製造工程において以下の条件に則っていることが重要な要件とされています。
➀文書化された計画に基づいている
②計画通りに実行されている
③実行したことを証明する記録を残している
④誰が見ても説明できる状態である
工程文書化の第一歩は、工程の目的と、境界(入力/出力)を構造化して明確にすることです。目的を達成するための条件や材料(入力)が決まれば、管理/記録しなければならない項目(出力)は自ずと明確になります。構造化することで他の工程でも活用が可能となります。
更に作業標準、工程パラメータとその管理幅(上限/下限)、設備・監視測定機器の条件および工程確認項目を加え、標準作業手順書(以下、SOP)として作成します。ここで重要なのは工程パラメータとその管理幅を明確にすることです。工程パラメータとその管理幅は品質のばらつきに直結します。SOPを基に実行されたことを証明するためには製造記録が不可欠です。
代表的な記録内容は以下です。
・製造日
・使用材料とロット(該当する場合は有効期限)
・作業者
・工程パラメータ(設定値/実測値)
・製造環境(温湿度、清浄度など)
・工程確認の結果(判定)
・使用設備(点検/校正状態や管理番号)
・管理番号(所定様式、改ざん防止)
記録の粒度は、各組織の品質マネジメントシステムに応じ決定されます。 記録は、製品の追跡(トレースフォワード)や遡及(トレースバック)、出荷ロットとの照合ができる状態にしておく必要があります。
そのためには、製造日や材料ロットだけでなく、作業者の教育訓練や力量評価、設備の点検・校正記録も含めて、まとめて管理しておくことが重要です。
医療機器の誤認を防ぐ表示のポイント
製品の識別は、「この製品が何であるか」だけではなく、「今どの状態にあるのか」までを、誰がどの工程でも理解できるように示す必要があります。製品名やロット番号などの表示だけでは不十分で、視覚的に分かる状態表示(検査前/後、作業中、出荷待ちなど)が必要です。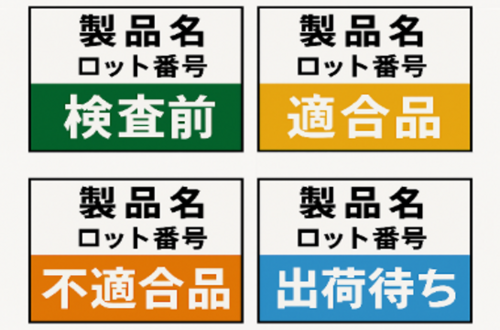 図1:状態表示の例
図1:状態表示の例
これは材料や仕掛品だけではありません。工程についても誰でも誤認することのないよう、状態表示が必要です。 図2:工程表示の例
図2:工程表示の例
表示についても各組織の品質マネジメントシステムにてその様式や色を定め、認識を統一しておきます。
また、システムを使用する場合は、システム上の情報との整合も必要となります。例えば、既に出荷しているものがいつまでも在庫として残っている、入荷したはずの部材がないなど、現物とシステムの両方が一致していると、誤認リスクは最小限に抑えられます。
品質を左右する医療機器製造での工程リスク管理とは
顧客所有物・保存(保管・取扱)・設備機器の管理は、本来それぞれ個別に管理される項目です。しかし、これらを「工程に与えるリスク」という視点で統一することで、管理するべきポイントが明確になります。
本章では、「工程に対して何がリスク源となり得るのか」という観点で整理します。
■顧客所有物(支給品、貸与品)
支給材料・貸与測定機器・図面・データなど、工程に投入される"外部起点"のインプットを指します。これらは工程の成立に直接影響するため、リスク源として扱います。
■保存条件(取扱、保管、防護)
製造工程を経た製品や仕掛品が、保管・取扱の間に規格や仕様から逸脱しないように管理する必要があります。劣化因子のリスクとして扱います。
■設備・監視測定機器
設備や測定機器は、工程が正しく成立するための基盤となります。校正や点検期限、警報履歴などを始動前点検項目として統合することで、リスクを最小限に抑えることが可能です。
工程に重大な影響を与えるリスクは、整理して可視化することで、影響度や優先度を明確にすることができます。
| リスク源 | 工程への影響例 | 必要な管理強度 | 記録(工程出力情報) |
| 顧客所有物 (支給材料・貸与測定器) |
材料規格不一致 →機能不良 貸与測定器の校正切れ →誤判定 |
・受入検査 ・ダブルチェック ・状態識別 ・顧客通知 |
・受入記録 ・受入検査記録 ・工程投入ロット記録 ・顧客通知の履歴 |
| 保存 (環境条件・取扱) |
無菌バリア劣化 光学部品変更 静電破壊 |
・差圧/清浄度/温湿度管理 ・クリーンルーム運用 ・除電対策 |
・環境モニタリング ・保管ロケーション記録 |
| 設備・測定機器 | 成立条件が満たされない →不具合流出 |
・バリデーション ・校正 ・保守点検 |
・バリデーション報告書 ・校正記録 ・稼働記録 |
工程の妥当性を証明する「プロセスバリデーション」とは
プロセスバリデーション(以下、PV)とは、後の検査だけでは工程の有効性を証明できない場合、事前に再現性を科学的根拠に基づき実証し、工程が管理された状態にすることです。医療機器では、
・射出成形
・洗浄
・無菌バリア包装
・滅菌
などの製造工程がその対象となります。
PVの"筋"を通すとは、「①工程評価(PV要否判断)→②プロトコール(計画)→③IQ/OQ/PQ*2→④維持管理→⑤再PV」の一連の流れが論理的につながっている状態を指します。
①工程評価
次の3要素のいずれかに該当する場合は必須です。
・検査で十分な適合性を確認できない(全数検査ができない)
・工程が不可逆的である(実証に破壊試験を伴う)
・患者リスクの高い重要工程である
②プロトコール(計画):事前に成立条件を明確にするために、以下の項目を含め文書化します。
・目的 ・統計的手法とサンプルサイズの根拠
・工程の範囲
・判定基準
・パラメータとその範囲
・逸脱時の扱い
③IQ/OQ/PQ*2
IQ:据付時適格性確認 設備が設計通りに据え付けられているかを確認します
OQ:運転時適格性確認 実際の製造条件を模倣し、上下限まで動作確認を行います
PQ:稼働性能適格性確認 通常運転条件下での再現性を確認します
④維持管理:バリデーションが成立している状態を維持します
管理図や工程能力指数などによって工程の安定状態を監視します
⑤再PV:工程変更時には変更の妥当性を評価し、場合によっては再PVを行います
まとめ
医療機器の製造工程における品質マネジメントは、単なる手順整備だけでなく、「再現性」と「説明可能性」をいかに担保するかが重要です。
しかし実際の現場では、「工程は整備されているものの運用にばらつきがある」「記録は残しているが十分に活用しきれていない」といった課題を抱えるケースも少なくありません。このような課題を解決するためには、工程設計から運用、改善までを一貫した視点で見直し、実務に即した形で仕組みを構築することが不可欠です。
鈴与では、医療機器製造業登録拠点およびISO13485に基づいた品質マネジメント体制のもと、製造工程の設計・運用から改善までを一貫してサポートしています。 工程の文書化、リスクマネジメント、プロセスバリデーションに至るまで、実運用に即した仕組みづくりをご提案可能です。
鈴与の医療機器物流サービスについて詳しく知りたい方は、こちらをご覧ください。
▶医療機器物流サービスページはこちら
また、現在の製造工程や品質管理体制に課題を感じている方は、ぜひ一度お気軽にご相談ください。
関連リンク



お問い合わせはこちら
Contact Us
関連記事
-

2026.03.12
医療機器の輸送・保管リスクと対策|設計・開発で見落としがちな物流リスクとは?
医療機器の輸送・保管には、温度・衝撃・荷役など多くの物流リスクが潜んでいます。本コラムでは、医療機器物流の全体像を整理し、設計・開発段階で見落とされが...
- 医療機器物流
-

2026.01.27
制度とリスクに強い医療機器物流へ|サプライチェーン再設計の重要性
現在、医療機器業界はUDI義務化の完全運用、在宅医療の急拡大、国際物流リスクなど大きな環境変化に直面しています。医療機器を扱う企業にとって、法規制への確実...
- 医療機器物流
-

2025.12.18
医療機器製造におけるリスクマネジメントとは
医療機器のリスクマネジメントとは、潜在的な危険を予測・評価し、受容可能範囲まで管理するとともに、市販後に顕在化した危害へ適切に対応する取り組みです。本...
- 医療機器物流